Testis tissue is very complex and heterogeneous as it is made up of several somatic (Leydig, Sertoli, peritubular myoid cells and others) and germ cell types (spermatogonia, spermatocytes and spermatids). Due to the high number of different cell types, histological and molecular studies have always been challenging. As an example, the use of bulk RNA sequencing techniques, has proven to be difficult to interpret as it is not possible to discern in which cell a specific gene is expressed. The recent advances in single cell sequencing technology have made it possible to study cell type-specific gene expression by measuring RNA abundance at a single cell level.
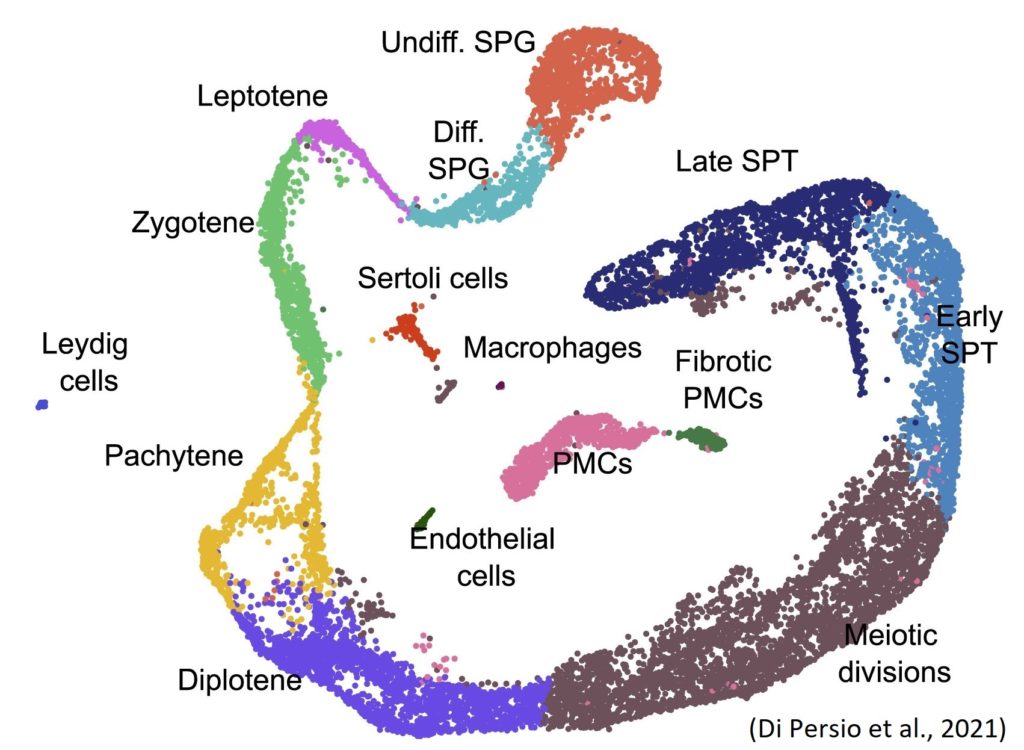
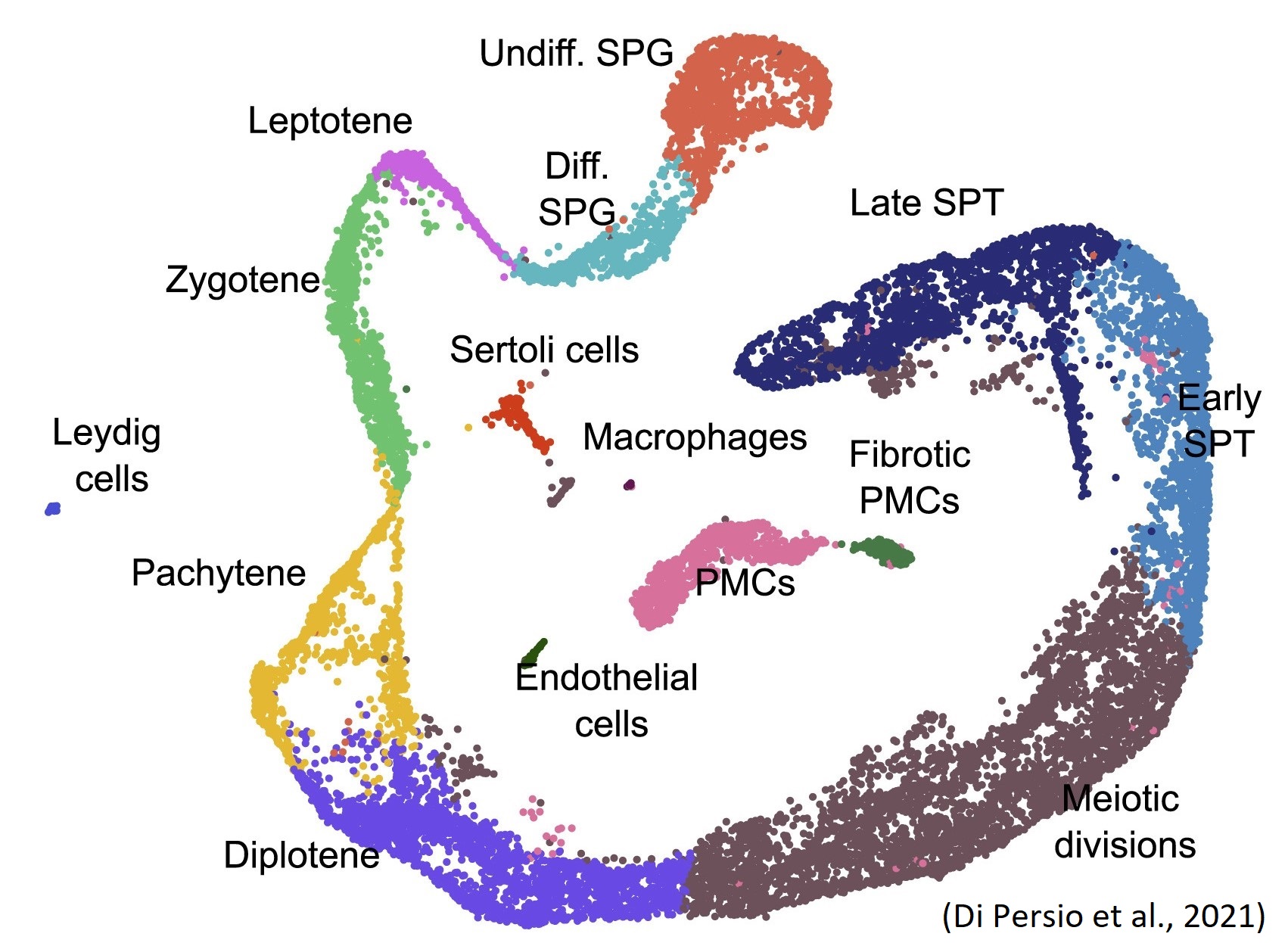
Figure 1 Uniform manifold approximation and projection (UMAP) plot showing different cell clusters.
Following this, the first adult human testis transcriptional cell atlases have been described by computationally reconstructing spermatogenesis and identifying new marker genes for each testicular cell type (Wang et al., 2018; Guo et al., 2018; Hermann et al., 2018, Sohni et al., 2019). Advanced single cell sequencing algorithms include pseudotime calculations, cell type identification and clustering analyses as new tools in fertility research. Different bioinformatic tools can also be accessed online. As an example, the ReproGenomics Viewer (https://rgv.genouest.org/), developed by the former NYRA board member Thomas Darde, or the Male Fertility Gene Atlas (https://mfga.uni-muenster.de/projectInfo.html), developed by Henrike Krenz (NYRA member), integrates OMICS data, allowing data visualization and interpretation in the context of other relevant datasets (Darde et al., 2019 and Krenz et al., 2020).
Sara Di Persio, who is also an active NYRA member, and colleagues recently performed single-cell RNA sequencing (Figure 1) on testicular cell suspensions obtained from testicular biopsies of men with normal and impaired spermatogenesis (cryptozoospermia – very low sperm number). As a result, they identified alterations in the spermatogonial stem cell compartment and specifically found 61 novel differentially expressed genes in the crypto undifferentiated spermatogonia. They further observed a transcriptional switch within the crypto spermatogonial compartment driven by increased and prolonged expression of the transcription factor EGR4. Consequently, EGR4 was suggested as a potential gatekeeper regulating the change in transcriptional profiles of spermatogonia in situations of impaired spermatogenesis (Di Persio et al., 2021).
References
M. Wang et al., Single-cell RNA sequencing analysis reveals sequential cell fate transition during human spermatogenesis, Cell Stem Cell, 23 (2018), pp. 599-614.e4
J. Guo et al., The adult human testis transcriptional cell atlas, Cell Res., 28 (2018), pp. 1141-1157
BP. Hermann et al., The Mammalian Spermatogenesis Single-Cell Transcriptome, from Spermatogonial Stem Cells to Spermatids, Cell Reports, Volume 25, Issue 6 (2018), pp. 1650-1667
A. Sohni et al., The Neonatal and Adult Human Testis Defined at the Single-Cell Level, Cell Reports, Volume 26, Issue 6 (2019), pp. 1501-1517
S. Di Persio et al., Single-cell RNA-seq unravels alterations of the human spermatogonial stem cell compartment in patients with impaired spermatogenesis. Cell Reports Medicine, 2(9) (2021), pp. 10395
T.A. Darde et al., The ReproGenomics Viewer: a multi-omics and cross-species resource compatible with single-cell studies for the reproductive science community, Bioinformatics 1;35(17) (2019), pp. 3133-3139
H. Krenz et al., The Male Fertility Gene Atlas: a web tool for collecting and integrating OMICS data in the context of male infertility, Hum Reprod 35(9)(2020), pp. 1983-1990